|
Epidemiology Diabetes Insipidus (DI) is a rare disorder caused by kidneys that cannot retain water (The Pituitary Foundation 2012). It is characterized by excessive thirst termed polydipsia and excretion of frequent volumes of hypotonic, diluted urine called polyuria (Khardori 2012). DI approximately affects 1 in 25,000 people in the UK (National Health Service 2012). Individuals of different age groups are susceptible to DI and majority are aged 10-20 (Makaryus 2006). The two main types of DI are Cranial DI (CDI) and Nephrogenic DI (NDI), CDI is more common than NDI and there is a worldwide tendency towards the growing CDI prevalence due to neurosurgical interventions, craniocerebral injuries and morbidity estimated at 30% (Thanh 2011). Pathology To understand the pathophysiological DI mechanisms, how water is normally balanced is addressed. Water metabolism and homeostasis are regulated by a balance between fluid intake that is controlled by thirst and output that is controlled by a nonapeptide hormone called arginine vasopressin (AVP) (Ahmed 2011:117). Both are regulated by the hypothalamus; a small gland located in the brain that interacts with pituitary gland to regulate hormonal secretion (Sehati n.d.). Water is distributed between two fluid compartments: intracellular (ICF) and extracellular (ECF). Dissolved constituents in each compartment affect its osmolality which defines as the number of osmotically-active particles in a solution (mOsm/kg) (Ahmed 2011:116). To maintain the gradient between both compartments Na/K-ATPase transmembrane enzyme performs ion exchange; sodium is transported from ICF to ECF and potassium vice versa using free energy gained from ATP hydrolysis (Ahmed 2011:116). The major stimulus in causing AVP release is when ECF osmolality is greater than ICF due to sweating or diarrhoea which causes the individual to become dehydrated. It increases pulse and decreases blood pressure, skin turgor, urine output and consciousness. The eyeballs become tender and the mucous membranes are dry (Smith 2012). High plasma osmolality is sensed by highly-sensitive osmoreceptors situated on the anterior hypothalamus that trigger the posterior pituitary lobe called neurohypophysis to increase AVP activation. AVP is synthesised in the neurosecretory cells, migrated via axonal transport to nerve endings, packaged into secretory vesicles with neurophysin II carrier protein and released into the systemic circulation pathway (Bowen 2006). Simultaneously, thirst is experienced because water deficiency activates the thirst centre located in the hypothalamus to increase fluid intake (Fursule 2006:17.10). When AVP reaches the kidneys it directs them to enhance water reabsorption. Water reabsorption is classified into obligatory and facultative depending on the site and nature. Obligatory reabsorption is the water quantity that is reabsorbed secondary to solute reabsorption in the proximal convoluted tubules. Facultative reabsorption is when AVP directs the remaining filtrate in distal and collecting tubules (Talwar 2006:284). The functions of AVP are mediated by three heptahelical vasopressin receptors: V1a, V1b and V2. The role of V1a is unknown whereas V1b mediates vasoconstriction, enhances prostaglandin synthesis and adrenocorticotropin release (Prasad 2001). V2 receptors are linked to heteromeric Gs proteins which are expressed on basolateral membrane cells in the cortical and medullary collecting ducts to increase water permeability. V2 receptors are unexpressed on apical membranes because these membranes are water impermeable in AVP absence and only permeable when water channels called aquaporins are present whereas basolateral membranes are constantly water permeable (Brandis n.d.). V2 receptors bind non-covalently to AVP which activates Gs protein and GDP is displaced by GTP. Gs dissociates into alpha (Gs) subunits and beta/gamma (β/γ) dimers. Gs subunit binds to adenyl cyclase to stimulate cAMP synthesis from ATP (Prasad 2001). This recruits a series of events that cause aquaporin 2-containing vesicles in the cytosol to move and attach to the apical membrane (Brandis n.d.). cAMP (secondary messenger) activates a holoenzyme called protein kinase A (PKA) which is targeted to many subcellular areas by PKA-anchoring proteins (AKAPs) (Klussmann 2001). Biochemical studies have shown that there are specific AKAP for each area. PKA entails a dimer of regulatory (R) subunits that is bound to catalytic (C) subunits (Klussmann 2001). The dimer mediates AKAP attachment to PKA and each cAMP molecule binds to a regulatory subunit to cause catalytic subunit (cPKA) to dissociate from PKA/AKAP complex and phosphorylate aquaporin 2 (AQP2) substrate; shown in Figure 1 (Klussmann 2001). 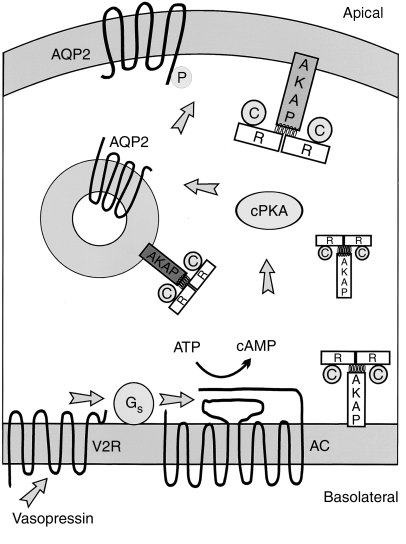 Figure 1: AKAP involvement in AQP2 translocation into apical membrane (Klussmann 2001) AQP2 inserts into apical membrane and forms a tetrameric complex with AVP and V2 receptor to increase water permeability. Water enters the cell through AQP2 in response to osmotic gradient and goes back into circulation across basolateral membrane (Brandis n.d.). Water reabsorption reduces plasma sodium concentration to permit sensitive feedback control of AVP secretion. Kidneys remove excess fluid from blood and store them in bladder as concentrated urine (Brandis n.d.). If ICF osmolality is greater than ECF i.e. overhydration can trigger vasopressin release. It is stimulated by high-pressure baroreceptors located in carotid sinus, that provide hypothalamic input via adrenergic pathways by responding to mean arterial blood pressure alterations (Fursule 2006:17.10). This consequently reduces AVP release; kidneys decrease plasma osmolality and increase urine osmolality to enhance diluted urine output to maintain water levels (British Society for Paediatric Endocrinology and Diabetes 2012). Other stimuli affect vasopressin for instance angiotensin-aldosterone system that primarily regulates body fluid shown in Figure 2 (Ahmed 2011:118). 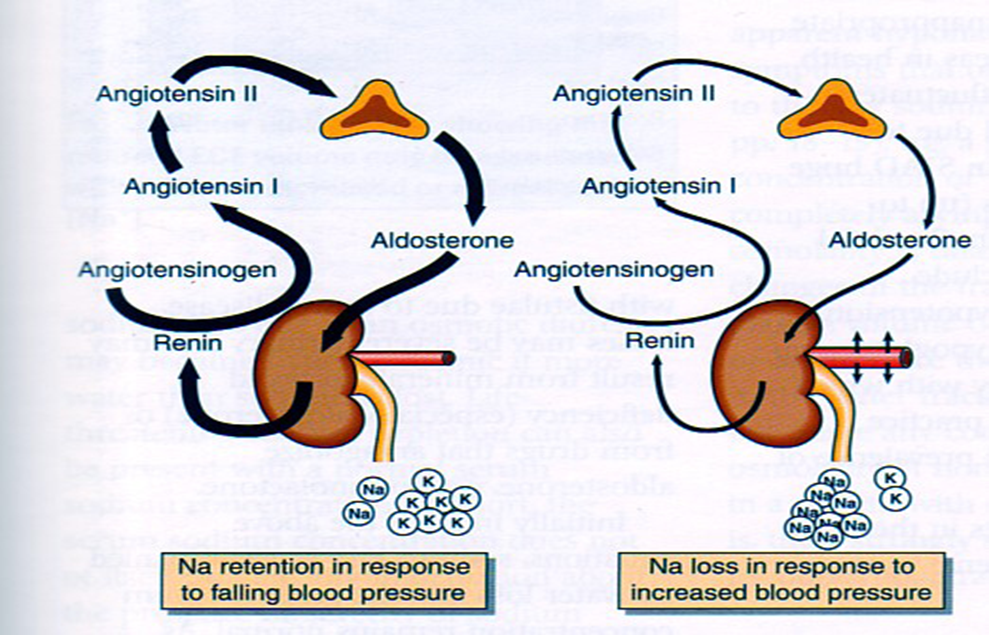 Figure 2: Angiotensin-aldosterone system (Smith 2012) CDI results from the impairment of ADH synthesis, transport and release. One of the main causes is damage or injury to the hypothalamo-neurohypophyseal region; it results from trauma, autoimmune diseases, infection such as meningitis and tuberculosis, surgery and benign or malignant tumours of the brain or pituitary (National Biotechnology Information 2012). Proximal injuries destruct neurons more than distal injuries in this sensitive region because in distal lesions only a few magnocellular neurons degenerate and regeneration of axon terminals by intact cell bodies takes weeks to months (Makaryus 2006).
Approximately, 30% of patients develop transient or permanent DI after pituitary surgery where the latter can be characterized by a triple-phase response (Humes 2001:752). At first, polyuria occurs for 1-2 days because AVP is decreased which lowers urine osmolality and increases urine output. Polydipsia also arises prohibiting the patient to concentrate urine. The second phase lasts for 2-3 days where vasopressin leaks from damaged AVP-secreting neurons which deregulates AVP release (Humes 2001:752). This can be linked to syndrome of inappropriate antiduiretic hormone hypersecretion (SIADH) as deregulated AVP secretion increases water reabsorption resulting in hyponatraemia, increased urine osmolality and decreased serum osmolality (Thomas 2012). The final phase is permanent DI where neuronal death impairs AVP synthesis (Humes 2001:752). However, post-operative polyuria does not always indicate DI as there is other causes for example osmotic diuresis that result from cerebral oedema treatment and removal of excess fluid given during surgery (Khardhori 2012). NDI occurs when high plasma osmolality cause the posterior pituitary to hyper-stimulate AVP but kidneys cannot respond to it. NDI is characterized by three dysfunctions: unmaintained corticomedullary osmotic gradients where water flows from collecting ducts into interstitial tissue, osmotic equilibration disorder due to cAMP defect between tubular contents and medullary interstitum. Thirdly, osmotic diuresis that rapidly flows tubular fluid preventing osmotic equilibration with medullary interstitium (Markayus 2006). Other DI types include Dipsogenic, caused by damage or defect to the thirst centre that leads to primary polydipsia which increases fluid intake to suppress AVP secretion and enhance urine output (Mangusan 2010). The exact lesion site is unknown although structural lesions can exist and can be conveyed in meningitis and multiple sclerosis (Khardori 2012). Gestational DI occurs during pregnancy where placental vasopressinase destructs AVP in the mother; pregnancy can magnify severity of existing NDI or CDI (Mangusan 2010). Aetiology Besides damage and injury to hypothalamo-neurohypophyseal region, there are idiopathic and familial CDI causes. Idiopathic CDI can develop when hypothalamic cells are damaged; the effect of inflammation and autoimmunity is becoming increasingly recognised and led to advances in imaging procedures and detection of antibodies that oppose AVP-secreting cells; reducing Idiopathic CDI incidence (Khardhori 2012). However, these antibodies can develop in Langerhans cell hisitiocytosis (LCH) patients which indicate it is an unreliable marker of autoimmune influence in CDI and can delay LCH diagnosis (Khardhori 2012). Most familial CDI cases are autosomal dominant inheritance caused by AVP neurophysin (AVP-NP2) gene defect on chromosome 20p13 that synthesises a mutant neurotoxic prohormone. Autosomal recessive cases are due to AVP-NP2 and WFS1 gene defects; WFS1 normally encodes wolframin, an endoplasmic reticular calcium channel (Kharkhodi 2012). NDI can result from systemic diseases and drugs that structurally or functionally alter kidneys’ ability to respond to AVP. For instance, polyuria, polydipsia and diuresis can be observed in hypokalaemic patients due to AVP-resistance in collecting ducts and alterations in generating and maintaining medullary osmotic gradient (Makaryus 2006). Drug-induced NDI is a side-effect of various drugs such as lithium where 60% of patients develop polydipsia and polyuria at the start of treatment (Makaryus 2006). Lithium increases prostaglandin levels, inhibits cAMP synthesis in collecting tubular cells and deregulates AQP2 (Getzinger 2007). 20-25% of lithium’s side effects are persistent even if levels return to normal (Makaryus 2006). Other drugs can induce NDI for example Gentamicin can impairs cellular response to AVP, colchicine inhibits cAMP by disrupting microtubules and recently forcarnet can cause NDI (Makaryus 2006). Familial NDI is rare and the two main genetic mutations are V2 receptor gene on chromosome Xq28 and AQP2 gene on chromosome 12q13. Studies have shown there are autosomal dominant and recessive inheritances of AQP2 mutations where AQP2 undergo conformational changes that effects fluid exchange in distal tubules and cause polyuria (Makaryus 2006). Diagnosis Polyuria, polydipsia, and electrolyte imbalance are the predominant DI manifestations (The Pituitary Foundation 2012). Laboratory tests are conducted to assess AVP secretion and confirm DI diagnosis. Urinalysis is performed to detect polyuria and differentiate from other polyuric causes. Serum electrolytes (sodium and potassium) are measured because they are elevated in DI. Blood glucose levels are analysed to exclude Diabetes mellitus as both have similar symptoms. Reference ranges of plasma and urine osmolality is 280-295 mOsm/kg and >750 mOsm/kg respectively whereas DI is characterized by high plasma osmolality <300 mOsm/kg with urine osmolality <750 mOsm/kg (Ahmed 2011:316). Water deprivation test examines the posterior pituitary function to distinguish between CDI and NDI by measuring changes in body weight, urine composition and output (Marshall 2008:44). The patient cannot drink fluid for 6-8 hours, urine and serum samples are collected for analysis; this test can be dangerous and close supervision is necessary. Normally with time, plasma osmolality is normal but urine osmolality increases (<800 mmol/kg) (Ahmed 2011:316). If plasma osmolality increases and the patient cannot concentrate his/her urine then a test using a synthetic AVP analogue called Desmopressin (DDVAP) is performed immediately. The patient sips water to decrease thirst and DDVAP is injected intramuscularly, urine specimens are collected and osmolality is measured (Beckett 2005:6). In CDI, desmopressin mimics AVP action causing concentrated urine; In NDI patients it is ineffective because kidneys are AVP-resistant. Desmopressin can also differentiate between two NDI defects: AQP2 responds to Desmopressin but V2 receptor cannot (Makaryus 2006). Other tests include MRI scanning of the brain and pituitary for possible DI causes (National Kidney and Urologic Diseases Information Clearinghouse 2012). Hypertonic saline infusion test is used to confirm CDI in polyuric patient that have normal plasma osmolality by comparing AVP response to plasma osmolality. NDI patients have normal AVP release in response to hyperosmolarity whereas CDI has low or no AVP rise (Barth 2001). Perinatal mutation testing is useful for early NDI diagnosis to prevent physical and mental retardation and repeated dehydration episodes (Bichet 2006). Treatment and Management of the disease DI treatment depends on the following factors: cause, age, history, extent of disease and expectations for the disease course. Drinking water corrects metabolic abnormalities that cause polyuria (National Health Service 2012). Desmopressin is the current drug choice that can be administered orally, nasally or parentally (Markayus 2006). It prevents water loss in kidneys to make less urine and respond less to fluid alterations. It is unprescribed for dipsogenic DI because it decreases urine output but not thirst and fluid intake which causes water intoxication that damages the brain. Most gestational DI cases are treated with desmopressin but not in rare cases of thirst mechanism abnormalities (Getzinger 2007). Thiazide diuretics for instance hydrochlorothiazide (HCTZ) combined with amiloride increases proximal absorption of sodium and water by reducing sodium and chloride distal absorption. Carbamazepine reduces sensitivity of ADH secretion but raises sensitivity in collecting duct for hydro-osmotic ADH action. Clofibrate is a lipid-lowering agent that stimulates ADH synthesis (Makaryus 2006). NDI is treated by correcting blood calcium and potassium levels and avoiding drugs that induce NDI enabling kidneys to respond to ADH; however prolonged lithium usage can be permanent. Current areas of Research Researchers are investigating cellular and molecular mechanisms that regulate body fluid to direct more effective treatments (National Kidney and Urologic Diseases Information Clearinghouse 2012). Gabbi et al. (2012) recently demonstrated oxysterol liver X receptor β (LXRβ) exhibits polyuria and polydipsia which decreases urine output and increases urine osmolality. LXRβ regulates water balance and will be useful to target treatments for water homeostatic disorders . A new assay was determined for copeptin, the c terminus of the vasopressin; it holds better specificity to diagnose DI after a recent study indicated limitations in current diagnostic biochemical tests (Fenske 2012).
1 Comment
|
This project began as a facebook page sharing information about different illnesses, diagnosis and treatments. We are now doing short articles :)
Health stuffArchives
April 2020
Categories |
 RSS Feed
RSS Feed
