|
Atherosclerosis is the development of atheromatous plaques in the inner arterial wall. It is the underlying reason for the pathophysiology and aetiology of many arterial diseases particularly coronary artery disease (CAD) where these plaques partially or completely block arteries. This limits the oxygenated blood supply to myocardium causing over 90,000 UK deaths a year despite rates decreasing since 1970s (BHF,2009). There are two main coronary arteries (right and left) arising from aortic sinus; the anatomy of coronary artery is shown in Figure 1a. 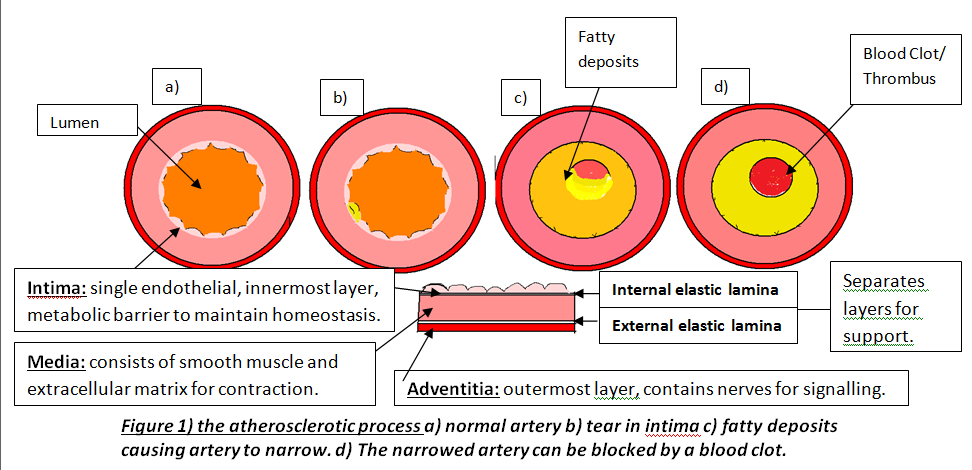
Atherosclerosis occurs in three major stages: fatty streak, plaque progression and disruption. Factors such as smoking cause endothelium to produce superoxide anions that interacts with other molecules. Superoxides are reactive oxygen species and free radicals of dioxygen due to unpaired electron (Widmaier,E. 2008:pp.84). This causes oxidative stress leading to endothelium dysfunction, allowing lipoproteins, calcium and fibrous tissues to enter and modify intima; Figure 1b. This initial damage increases CAD risk and initiates leukocyte recruitment primarily monocytes and T lymphocytes to enter inflammatory site. Monocytes differentiate into macrophages and secrete growth factors, platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF- β), and have LDL-receptors to engulf lipids forming foam cells (Lilly,L. 2007 pp.126). These foam cells accumulate to form yellowish fatty streaks; Figure 1c. Plaque growth progresses when smooth muscle migrates to intima. PDGF allows smooth muscle to proliferate for collagen synthesis whereas TGF- β stimulate the process losing the arterial wall’s elasticity (Lilly,L. 2007:pp.127). A fibrous cap forms protecting atheroma, causing an ischaemic condition called angina. Over decades, dispute occurs between growth factors and IFN-γ factor; an interferon released by leukocytes inhibiting collagen synthesis. Disruption occurs when plaques rupture triggering a coagulation cascade where thrombus and fibrin deposit in arterial wall further reducing lumen and cause coronary occlusion; Figure 1d. This can lead to myocardial infarction (MI) where inadequate blood supply to part of the heart causes myocardial necrosis and can be fatal. Other causes of CAD are risk factors that promote atherogenesis. Controllable factors include diet, exercise and obesity. Obesity increases risk because abdominal fat enhances low-density lipoproteins (LDL) production and heart workload. Studies show that a third of CAD patients in developed countries are due to obesity emphasising its impact (BHF, 2008). As well as being an independent factor, it can trigger other factors, for instance, cholesterol where high LDL and triglyceride levels inside coronary arteries increase plaque formation. Low high-density lipoproteins (HDL) levels raises risk because epidemiological studies show that high HDL levels enhances endothelium function by preventing cholesterol entry (Brubaker,P. 2002:pp.9). There are also uncontrollable factors that contribute to CAD, for instance, gender and age. Women have higher HDL2 levels, a more cholesterol rich, than men preventing cholesterol entry and lowering CAD risk. Studies show 40% of deaths in 65-74 years old are due to CAD because myocardium cardiac function has decelerated, myocardium becomes less efficient and cardiac muscle (BJN, 2009). Individuals at risk of CAD are diagnosed using different examinations after experiencing symptoms such as chest discomfort where it spreads to arms and other areas; MI is identified when discomfort is beyond 15 minutes (Julian,D. 2005:pp.116). Clinical history and electrocardiogram (ECG) is needed because ECG records heart’s electrical activity by placing electrodes on arms, legs and chests detecting characteristic changes. For instance, ST segment lowers when there is myocardial ischaemia but elevates when MI initiates; Figure 2. 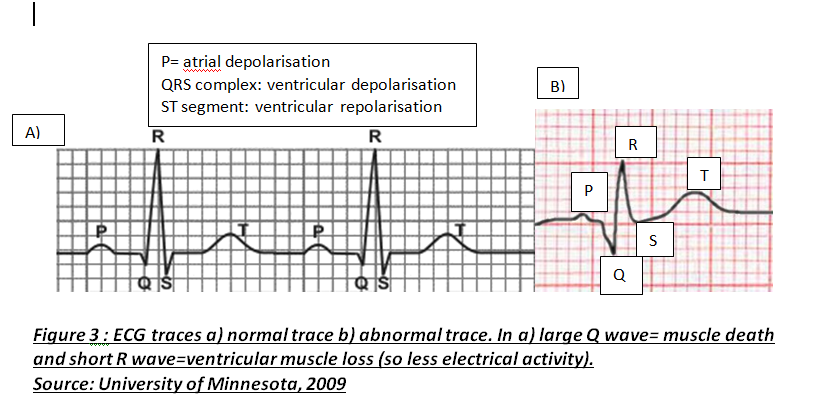
Another diagnosis is cardiac markers where high enzyme concentrations in cardiac tissues indicate myocardium necrosis. Tropinin T and I regulate cardiac muscle contraction and are mainly tested because they are sensitive and raise their serum activity when they detect any muscle injury (Julian,D. 2005:pp.112).
Treatment of CAD aims to improve coronary circulation by dilating coronary arteries which lowers pressure and myocardial oxygen demand and this reduces heart work. Drugs like nitrates have this pharmacological effect by relaxing smooth muscle whereas other drugs like atorvastatin lower LDL cholesterol by inhibiting HMG-CoA reductase; enzyme responsible for cholesterol synthesis (Kendall,M. 1998:pp.109). However, atorvastatin can cause myositis which affects muscles emphasising how medications can cause side-effects. If medications fail to work, surgery is needed. Coronary artery bypass grafting (CABG) involves using a vessel, normally saphenous vein, which bypass narrowed arteries providing an alternative route for blood flow. In 2002, mortality rate for Coronary Artery Bypass operations was 1.8% highlighting how surgery is effective (Barrett,D. 2006:pp.212). However, studies show that preventing modifiable factors by lifestyle adjustments are more effective than other treatments. Stopping smoking, maintaining healthy weight by exercising and eating low-saturated fat diet helps reduce cholesterol, blood pressure and controls glucose levels if diabetic. Ultimately, researchers are investigating possible new factors and cardioprotective drugs which emphasises the epidemic’s extent.
0 Comments
Advanced understanding in the molecular aspect of disease generated progress leading to a novel healthcare approach called stratified medicine. From recent years it transitioned from several stratified medicines such as Imanitib, a mutant kinase inhibitor prescribed for patients with chronic myeloid leukaemia to a variety of medicines today such as Zelboraf; targets BRAF proteins in patients with V600E mutation in melanoma. Stratified medicine is defined as a treatment derived from utilizing molecular or genetic information to discover patients with different mechanisms of disease or a specific therapeutic response as illustrated in Figure 1. This allows identification and development of therapies and diagnostic tools that give better health stances such as safety and effectiveness, for an aimed sub-group of patients that have analogous biological characteristics. Besides patients, there are also benefits for clinicians, economics and healthcare system. Thus, stratification plays a significant role in medicine especially cancer therapy. 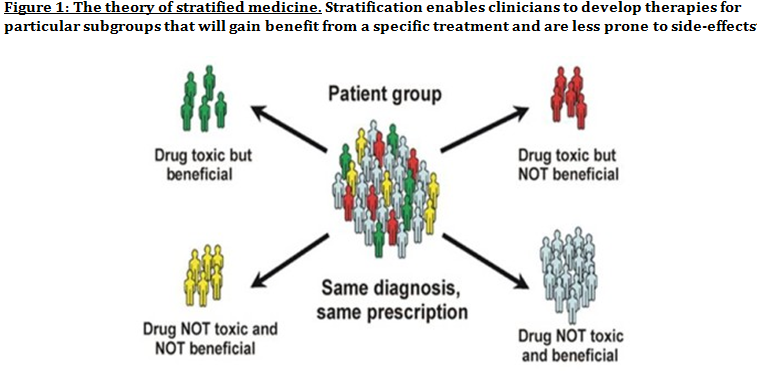 One of the ways in which stratified medicine alters cancer treatment can be seen through HER-2 status in breast cancer management illustrated in Figure 2. This is where findings from prognosis research are translated towards developing better patient outcomes. At first, HER-2 protein was found as a prognostic factor due to overexpression in breast cancer cells. This led to development of Trastuzumab (Herceptin) as a potential treatment against HER-2. Then, evaluation in trials took place where subjects recruited were women with HER-2 positive cancers. Due to successful results, Herceptin was prescribed to HER-2 positive patients and not those that were HER-2 negative. Those that were negative had alternative treatments. 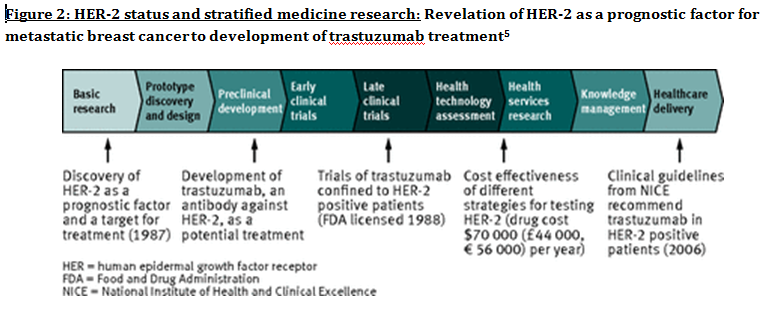 Another way in which stratified medicine is altering cancer treatment is via biomarkers. Biomarkers are defined as biological characteristics that give an indication of pathogenic process, normal biological state or pharmacological response to a therapeutic involvement. They can be cellular, biochemical or genetics. These markers allow identification of sub-groups that are able to respond positively or negatively due to a particular treatment. A number of randomised control designs (RCT) are used to assess predictive biomarkers such as biomarker-strategy design presented in Figure 3A. They are utilized in scenarios where there are two or more therapeutic options8. Patients are assigned on a random basis despite of their biomarker status8. An analysis plan is then stratified via biomarker to examine the treatment effect. An example of this is Marker Validation for Erlotinib in Lung Cancer (MARVEL) trial8. Patients with non-small cell lung cancer (NSCLC) are allocated to either treatment: Pemetrexed or Erlotinib. The analysis plan is then stratified via epidermal growth factor receptor gene (EGFR) status. This is followed by qualitative analysis via fluorescent in situ hybridization (FISH). Advantages of using biomarker-strategy design are that it examines relative efficacy of each sub-group. However, it is unreasonable for multifaceted therapeutic approaches and several treatments maybe inappropriate for some biomarker groups. Another type of RCT design is Enrichment design (Figure 3B), it consists of evaluating all patients, however the analysis plan is stratified to specific patients with definite biomarker values8. For example in CALGB-10603 trial, utilized a predictive biomarker allowing only acute myeloid leukaemia patients with FLT3 mutation. They were allocated randomly to a normal treatment or a treatment that entailed FLT3 kinase inhibitor; midostaurin, whereas, those without FLT3 mutation where ‘off-study’. One of the benefits of using enrichment design is that it utilizes a small sample. Nevertheless, it requires potent biological basis and the biomarker may not identify correctly the sub-group that would gain. Additionally, biomarker-strategy design (Figure 3C) entails patients who are allocated randomly and uses an investigational treatment arm that utilizes biomarker to direct treatment or to a control arm that does not direct treatment8. An example of this can be seen with Excision repair cross-complementing 1 (ERCC1) as biomarker. It is linked to cisplatin resistance in NSCLC. Therefore, in the trial patients are allocated on a random basis to the control arm8. This is where they will be given two treatments: docetaxel and cisplatin8. However patients may be allocated to the biomarker-strategy arm where patients are altered to gemcitabine and docetaxel treatment rather than being categorized as cisplatin resistant. Patients thare are not resistant to cisplatin undergo the standard treatment which is composed of cisplatin and docetaxel. This design can be used in complex therapeutic approaches. On the contrary, a positive trial does not demonstrate biomarker use. This emphasises the clinical relevance of biomarkers towards cancer treatment. However, there are disadvantages of utilizing biomarkers such as insufficient handling, processing and storage of samples. This can lead to their degradation and false results. For improvement, removal of quality samples for biomarker analysis from reliable sources reduces issues to patients. This will need design and validation of protocols for procedures. 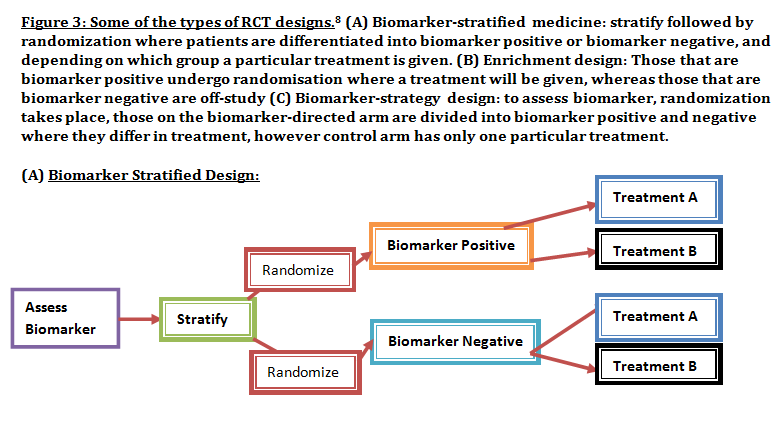 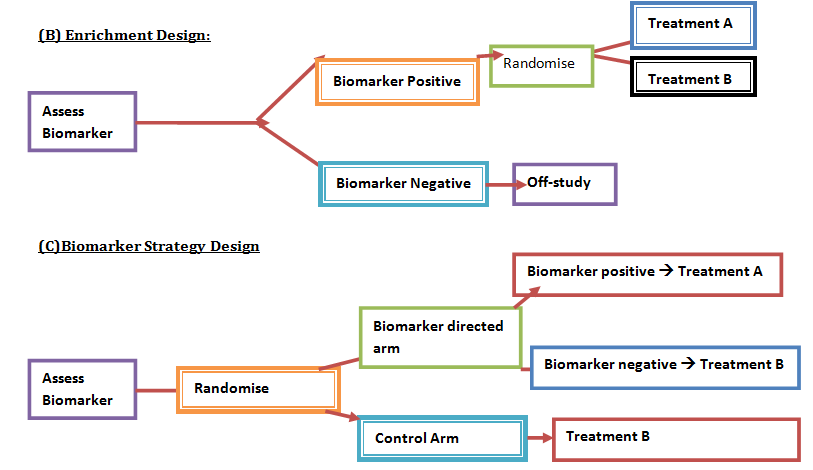 A technique in which cancers can be stratified is genomic analysis using DNA and RNA microarrays. Microarrays consist of RNA or DNA pieces from genes and quantify the amount of corresponding DNA or RNA in the provided sample. Therefore, genetic variants or expression levels of disease-related genes can be observed. For example MammaPrint, a microarray-based diagnostic technique can examine genetic variants in breast cancer tissue to estimate possibility of metastasis. However, microarrays are limited because they can only assess genes presented there. DNA microarrays will potentially be substituted with whole genome sequencing due to benefit of obtaining complete information, whereas RNA microarrays remains valuable for exploring gene expression. Moreover, metabolomic and protein analyses are another method used to stratify patients. Microarrays can be utilized where antibodies fuse to desired metabolite or protein variant. In comparison to genomics, both analyses provide more information on molecular mechanisms involved in a disease. A combination of their data, genomics and clinical outcomes indicates the impact of biomedical and health informatics on progress in stratified medicine. Nevertheless, storage conditions can affect results despite extraction of molecules can be easily obtained. Other techniques by which cancers maybe stratified are immunohistochemistry and polymerase chain reaction (PCR). They are routinely used in contrast to genomic, proteomic or metabolomic analysis. Immunohistochemistry utilizes antibodies that bind to a specific molecule. This is normally a cell-surface protein and appears with a unique colour under microscopic analysis. This technique can be performed using oestrogen receptor biomarker in breast cancer. Another example is PD-1 ligand in directing antibody therapy for patients with colorectal cancer and melanoma. Nonetheless, immunohistochemistry performed in clinical setting, diagnostic-grade antibodies are not used always. In relation to qPCR, they examine a desired DNA sequence in the sample that are normally cancer mutations. Similar to immunohistochemistry, they do not always perform using standardised processes. Thus, prospective techniques such as X-ray computed tomography, positron-emission tomography (PET) and magnetic resonance imaging do not require invasive procedures to obtain clinical samples. To observe the patient’s interior body, one examines what happens between electromagnetic radiations with the body. 18-fluorodeoxyglucose (FDG), a PET-active glucose analogue that resides in malignant tumour tissues with enhanced metabolic rate. In combination with PET, FDG is useful for diagnosis and examining therapeutic response in many types of cancer such as lymphomas. From a bedside perspective, molecular profiling appears to be a potential technique in identifying biomarkers in renal cell carcinoma aiding in decision-making for kidney cancer patients illustrated in Figure 5. Molecular profile is an analysis of protein and gene expression and leads to developing novel targeted therapeutic response. 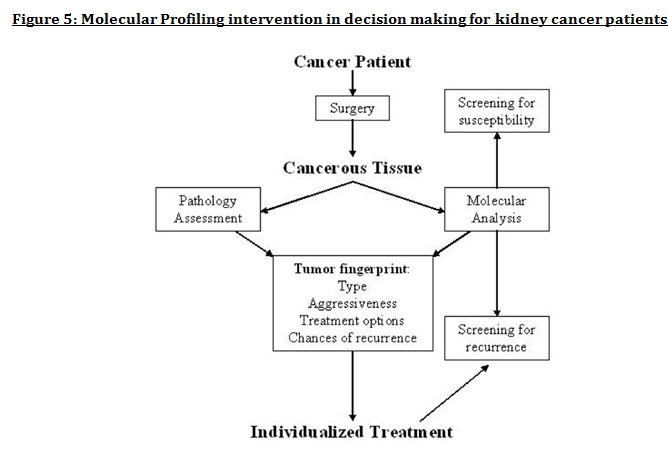 Furthermore, mutational stratification aids clinicians to determine which individuals will respond to which particular treatment. For instance, identifying KRAS in bowel cancer, Cetuximab and Panitumumab are observed to see if it implicated. Another example of how mutational stratification is likely to alter cancer treatment is gastrointestinal stromal tumour(GIST). Patients with GIST are prescribed with Imatinib. Some patients benefited where there was low GIST recurrence rates whereas others did not. Recent studies have demonstrated how mutational analysis is a predictive substance of recurrence-free survival. Patients with mutations in exon 11 of KIT gene, had more recurrence-free survival in comparison to patients with exon 9. Patients with wild-type GIST generally had disease but narrow sensitivity to Imatinib in most cases. Thus, risk-stratification and mutational analysis is necessary for optimising therapeutic approaches.
Lung cancer is another example of where mutational stratification has been used. It is the most common cause of mortality globally. According to statistics, 70% of NSCLC patients have durable effect and limited treatment obtained where 50% of them have an unknown cause. By using a specific genetic dependency screen one would be able to recognise vital somatic mutations. Findings discovered 3 kinases that have gain-in-function mutations in lung cancer mediating towards ERK pathway: PAK5, FGFR4 and MAP3K9. Consequently, mutated kinases prevent proliferation, inhibit downstream signalling events and eradicate lung cancer cells. Ultimately, stratified medicine plays a significant role in cancer treatment. Researchers are currently identifying more biomarkers to improve potential treatments. Cancer Research UK’s Stratified Medicine Programme entails molecular basis information of 9,000 tumours. It is co-working with pharmaceutical companies such as Pfizer to develop a national service model to routinely examine tumours for particular genetic variations. This will help clinicians with decision–making and aid researchers on how particular variations influence specific treatments. Epidemiology Diabetes Insipidus (DI) is a rare disorder caused by kidneys that cannot retain water (The Pituitary Foundation 2012). It is characterized by excessive thirst termed polydipsia and excretion of frequent volumes of hypotonic, diluted urine called polyuria (Khardori 2012). DI approximately affects 1 in 25,000 people in the UK (National Health Service 2012). Individuals of different age groups are susceptible to DI and majority are aged 10-20 (Makaryus 2006). The two main types of DI are Cranial DI (CDI) and Nephrogenic DI (NDI), CDI is more common than NDI and there is a worldwide tendency towards the growing CDI prevalence due to neurosurgical interventions, craniocerebral injuries and morbidity estimated at 30% (Thanh 2011). Pathology To understand the pathophysiological DI mechanisms, how water is normally balanced is addressed. Water metabolism and homeostasis are regulated by a balance between fluid intake that is controlled by thirst and output that is controlled by a nonapeptide hormone called arginine vasopressin (AVP) (Ahmed 2011:117). Both are regulated by the hypothalamus; a small gland located in the brain that interacts with pituitary gland to regulate hormonal secretion (Sehati n.d.). Water is distributed between two fluid compartments: intracellular (ICF) and extracellular (ECF). Dissolved constituents in each compartment affect its osmolality which defines as the number of osmotically-active particles in a solution (mOsm/kg) (Ahmed 2011:116). To maintain the gradient between both compartments Na/K-ATPase transmembrane enzyme performs ion exchange; sodium is transported from ICF to ECF and potassium vice versa using free energy gained from ATP hydrolysis (Ahmed 2011:116). The major stimulus in causing AVP release is when ECF osmolality is greater than ICF due to sweating or diarrhoea which causes the individual to become dehydrated. It increases pulse and decreases blood pressure, skin turgor, urine output and consciousness. The eyeballs become tender and the mucous membranes are dry (Smith 2012). High plasma osmolality is sensed by highly-sensitive osmoreceptors situated on the anterior hypothalamus that trigger the posterior pituitary lobe called neurohypophysis to increase AVP activation. AVP is synthesised in the neurosecretory cells, migrated via axonal transport to nerve endings, packaged into secretory vesicles with neurophysin II carrier protein and released into the systemic circulation pathway (Bowen 2006). Simultaneously, thirst is experienced because water deficiency activates the thirst centre located in the hypothalamus to increase fluid intake (Fursule 2006:17.10). When AVP reaches the kidneys it directs them to enhance water reabsorption. Water reabsorption is classified into obligatory and facultative depending on the site and nature. Obligatory reabsorption is the water quantity that is reabsorbed secondary to solute reabsorption in the proximal convoluted tubules. Facultative reabsorption is when AVP directs the remaining filtrate in distal and collecting tubules (Talwar 2006:284). The functions of AVP are mediated by three heptahelical vasopressin receptors: V1a, V1b and V2. The role of V1a is unknown whereas V1b mediates vasoconstriction, enhances prostaglandin synthesis and adrenocorticotropin release (Prasad 2001). V2 receptors are linked to heteromeric Gs proteins which are expressed on basolateral membrane cells in the cortical and medullary collecting ducts to increase water permeability. V2 receptors are unexpressed on apical membranes because these membranes are water impermeable in AVP absence and only permeable when water channels called aquaporins are present whereas basolateral membranes are constantly water permeable (Brandis n.d.). V2 receptors bind non-covalently to AVP which activates Gs protein and GDP is displaced by GTP. Gs dissociates into alpha (Gs) subunits and beta/gamma (β/γ) dimers. Gs subunit binds to adenyl cyclase to stimulate cAMP synthesis from ATP (Prasad 2001). This recruits a series of events that cause aquaporin 2-containing vesicles in the cytosol to move and attach to the apical membrane (Brandis n.d.). cAMP (secondary messenger) activates a holoenzyme called protein kinase A (PKA) which is targeted to many subcellular areas by PKA-anchoring proteins (AKAPs) (Klussmann 2001). Biochemical studies have shown that there are specific AKAP for each area. PKA entails a dimer of regulatory (R) subunits that is bound to catalytic (C) subunits (Klussmann 2001). The dimer mediates AKAP attachment to PKA and each cAMP molecule binds to a regulatory subunit to cause catalytic subunit (cPKA) to dissociate from PKA/AKAP complex and phosphorylate aquaporin 2 (AQP2) substrate; shown in Figure 1 (Klussmann 2001). 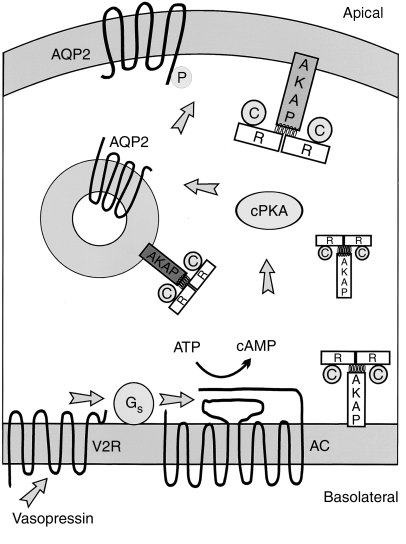 Figure 1: AKAP involvement in AQP2 translocation into apical membrane (Klussmann 2001) AQP2 inserts into apical membrane and forms a tetrameric complex with AVP and V2 receptor to increase water permeability. Water enters the cell through AQP2 in response to osmotic gradient and goes back into circulation across basolateral membrane (Brandis n.d.). Water reabsorption reduces plasma sodium concentration to permit sensitive feedback control of AVP secretion. Kidneys remove excess fluid from blood and store them in bladder as concentrated urine (Brandis n.d.). If ICF osmolality is greater than ECF i.e. overhydration can trigger vasopressin release. It is stimulated by high-pressure baroreceptors located in carotid sinus, that provide hypothalamic input via adrenergic pathways by responding to mean arterial blood pressure alterations (Fursule 2006:17.10). This consequently reduces AVP release; kidneys decrease plasma osmolality and increase urine osmolality to enhance diluted urine output to maintain water levels (British Society for Paediatric Endocrinology and Diabetes 2012). Other stimuli affect vasopressin for instance angiotensin-aldosterone system that primarily regulates body fluid shown in Figure 2 (Ahmed 2011:118). 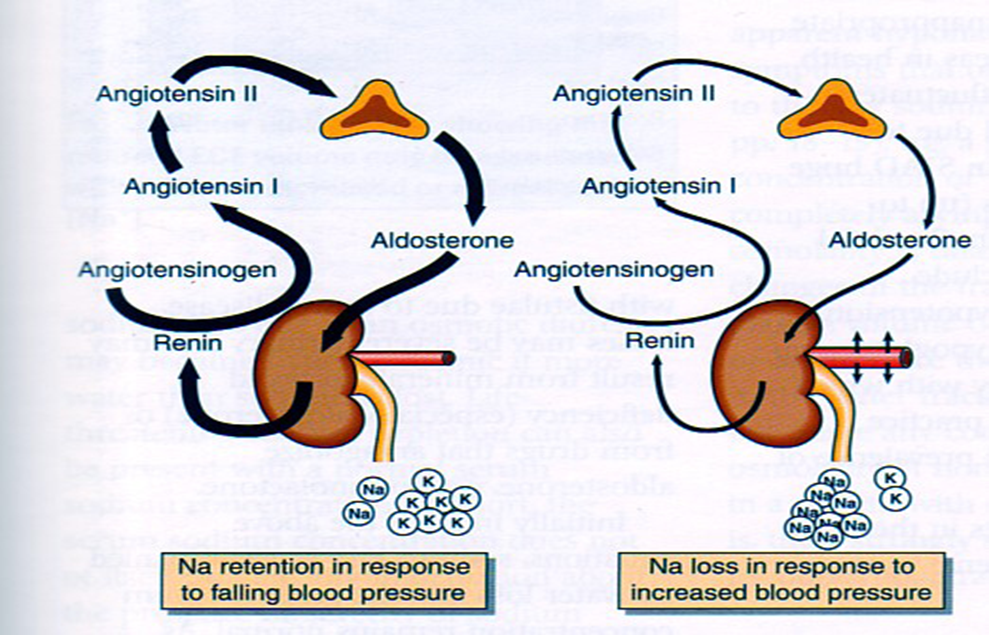 Figure 2: Angiotensin-aldosterone system (Smith 2012) CDI results from the impairment of ADH synthesis, transport and release. One of the main causes is damage or injury to the hypothalamo-neurohypophyseal region; it results from trauma, autoimmune diseases, infection such as meningitis and tuberculosis, surgery and benign or malignant tumours of the brain or pituitary (National Biotechnology Information 2012). Proximal injuries destruct neurons more than distal injuries in this sensitive region because in distal lesions only a few magnocellular neurons degenerate and regeneration of axon terminals by intact cell bodies takes weeks to months (Makaryus 2006).
Approximately, 30% of patients develop transient or permanent DI after pituitary surgery where the latter can be characterized by a triple-phase response (Humes 2001:752). At first, polyuria occurs for 1-2 days because AVP is decreased which lowers urine osmolality and increases urine output. Polydipsia also arises prohibiting the patient to concentrate urine. The second phase lasts for 2-3 days where vasopressin leaks from damaged AVP-secreting neurons which deregulates AVP release (Humes 2001:752). This can be linked to syndrome of inappropriate antiduiretic hormone hypersecretion (SIADH) as deregulated AVP secretion increases water reabsorption resulting in hyponatraemia, increased urine osmolality and decreased serum osmolality (Thomas 2012). The final phase is permanent DI where neuronal death impairs AVP synthesis (Humes 2001:752). However, post-operative polyuria does not always indicate DI as there is other causes for example osmotic diuresis that result from cerebral oedema treatment and removal of excess fluid given during surgery (Khardhori 2012). NDI occurs when high plasma osmolality cause the posterior pituitary to hyper-stimulate AVP but kidneys cannot respond to it. NDI is characterized by three dysfunctions: unmaintained corticomedullary osmotic gradients where water flows from collecting ducts into interstitial tissue, osmotic equilibration disorder due to cAMP defect between tubular contents and medullary interstitum. Thirdly, osmotic diuresis that rapidly flows tubular fluid preventing osmotic equilibration with medullary interstitium (Markayus 2006). Other DI types include Dipsogenic, caused by damage or defect to the thirst centre that leads to primary polydipsia which increases fluid intake to suppress AVP secretion and enhance urine output (Mangusan 2010). The exact lesion site is unknown although structural lesions can exist and can be conveyed in meningitis and multiple sclerosis (Khardori 2012). Gestational DI occurs during pregnancy where placental vasopressinase destructs AVP in the mother; pregnancy can magnify severity of existing NDI or CDI (Mangusan 2010). Aetiology Besides damage and injury to hypothalamo-neurohypophyseal region, there are idiopathic and familial CDI causes. Idiopathic CDI can develop when hypothalamic cells are damaged; the effect of inflammation and autoimmunity is becoming increasingly recognised and led to advances in imaging procedures and detection of antibodies that oppose AVP-secreting cells; reducing Idiopathic CDI incidence (Khardhori 2012). However, these antibodies can develop in Langerhans cell hisitiocytosis (LCH) patients which indicate it is an unreliable marker of autoimmune influence in CDI and can delay LCH diagnosis (Khardhori 2012). Most familial CDI cases are autosomal dominant inheritance caused by AVP neurophysin (AVP-NP2) gene defect on chromosome 20p13 that synthesises a mutant neurotoxic prohormone. Autosomal recessive cases are due to AVP-NP2 and WFS1 gene defects; WFS1 normally encodes wolframin, an endoplasmic reticular calcium channel (Kharkhodi 2012). NDI can result from systemic diseases and drugs that structurally or functionally alter kidneys’ ability to respond to AVP. For instance, polyuria, polydipsia and diuresis can be observed in hypokalaemic patients due to AVP-resistance in collecting ducts and alterations in generating and maintaining medullary osmotic gradient (Makaryus 2006). Drug-induced NDI is a side-effect of various drugs such as lithium where 60% of patients develop polydipsia and polyuria at the start of treatment (Makaryus 2006). Lithium increases prostaglandin levels, inhibits cAMP synthesis in collecting tubular cells and deregulates AQP2 (Getzinger 2007). 20-25% of lithium’s side effects are persistent even if levels return to normal (Makaryus 2006). Other drugs can induce NDI for example Gentamicin can impairs cellular response to AVP, colchicine inhibits cAMP by disrupting microtubules and recently forcarnet can cause NDI (Makaryus 2006). Familial NDI is rare and the two main genetic mutations are V2 receptor gene on chromosome Xq28 and AQP2 gene on chromosome 12q13. Studies have shown there are autosomal dominant and recessive inheritances of AQP2 mutations where AQP2 undergo conformational changes that effects fluid exchange in distal tubules and cause polyuria (Makaryus 2006). Diagnosis Polyuria, polydipsia, and electrolyte imbalance are the predominant DI manifestations (The Pituitary Foundation 2012). Laboratory tests are conducted to assess AVP secretion and confirm DI diagnosis. Urinalysis is performed to detect polyuria and differentiate from other polyuric causes. Serum electrolytes (sodium and potassium) are measured because they are elevated in DI. Blood glucose levels are analysed to exclude Diabetes mellitus as both have similar symptoms. Reference ranges of plasma and urine osmolality is 280-295 mOsm/kg and >750 mOsm/kg respectively whereas DI is characterized by high plasma osmolality <300 mOsm/kg with urine osmolality <750 mOsm/kg (Ahmed 2011:316). Water deprivation test examines the posterior pituitary function to distinguish between CDI and NDI by measuring changes in body weight, urine composition and output (Marshall 2008:44). The patient cannot drink fluid for 6-8 hours, urine and serum samples are collected for analysis; this test can be dangerous and close supervision is necessary. Normally with time, plasma osmolality is normal but urine osmolality increases (<800 mmol/kg) (Ahmed 2011:316). If plasma osmolality increases and the patient cannot concentrate his/her urine then a test using a synthetic AVP analogue called Desmopressin (DDVAP) is performed immediately. The patient sips water to decrease thirst and DDVAP is injected intramuscularly, urine specimens are collected and osmolality is measured (Beckett 2005:6). In CDI, desmopressin mimics AVP action causing concentrated urine; In NDI patients it is ineffective because kidneys are AVP-resistant. Desmopressin can also differentiate between two NDI defects: AQP2 responds to Desmopressin but V2 receptor cannot (Makaryus 2006). Other tests include MRI scanning of the brain and pituitary for possible DI causes (National Kidney and Urologic Diseases Information Clearinghouse 2012). Hypertonic saline infusion test is used to confirm CDI in polyuric patient that have normal plasma osmolality by comparing AVP response to plasma osmolality. NDI patients have normal AVP release in response to hyperosmolarity whereas CDI has low or no AVP rise (Barth 2001). Perinatal mutation testing is useful for early NDI diagnosis to prevent physical and mental retardation and repeated dehydration episodes (Bichet 2006). Treatment and Management of the disease DI treatment depends on the following factors: cause, age, history, extent of disease and expectations for the disease course. Drinking water corrects metabolic abnormalities that cause polyuria (National Health Service 2012). Desmopressin is the current drug choice that can be administered orally, nasally or parentally (Markayus 2006). It prevents water loss in kidneys to make less urine and respond less to fluid alterations. It is unprescribed for dipsogenic DI because it decreases urine output but not thirst and fluid intake which causes water intoxication that damages the brain. Most gestational DI cases are treated with desmopressin but not in rare cases of thirst mechanism abnormalities (Getzinger 2007). Thiazide diuretics for instance hydrochlorothiazide (HCTZ) combined with amiloride increases proximal absorption of sodium and water by reducing sodium and chloride distal absorption. Carbamazepine reduces sensitivity of ADH secretion but raises sensitivity in collecting duct for hydro-osmotic ADH action. Clofibrate is a lipid-lowering agent that stimulates ADH synthesis (Makaryus 2006). NDI is treated by correcting blood calcium and potassium levels and avoiding drugs that induce NDI enabling kidneys to respond to ADH; however prolonged lithium usage can be permanent. Current areas of Research Researchers are investigating cellular and molecular mechanisms that regulate body fluid to direct more effective treatments (National Kidney and Urologic Diseases Information Clearinghouse 2012). Gabbi et al. (2012) recently demonstrated oxysterol liver X receptor β (LXRβ) exhibits polyuria and polydipsia which decreases urine output and increases urine osmolality. LXRβ regulates water balance and will be useful to target treatments for water homeostatic disorders . A new assay was determined for copeptin, the c terminus of the vasopressin; it holds better specificity to diagnose DI after a recent study indicated limitations in current diagnostic biochemical tests (Fenske 2012). What is Obesity?Obesity is a condition where a person has an abnormal high triglyceride amount stored in adipocytes (fat cells) under the skin. Fulurija et al. reported it being one of the leading global health issues that result from a combination of genetic susceptibility, lack of exercise and high-energy food that are becoming increasingly available. Recent epidemiological studies suggest there is a causative relationship between obesity and the incidence of certain cancers including melanoma. However, the mechanism of melanoma is unknown despite obesity’s effect in progression of other cancers such as breast, colon and liver have been widely investigated (Pandey et al.) What is malignant melanoma? 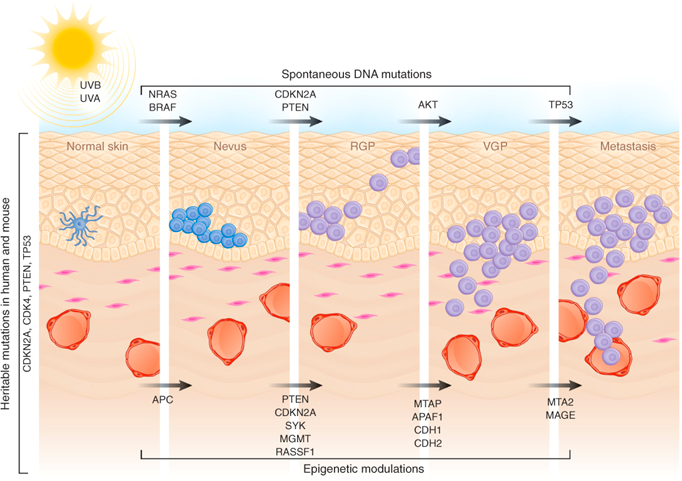
Malignant melanoma (MM) is a rare skin cancer that occurs in the following stages: Radial growth phase (RGP): Multiple clusters of abnormal melanocytes termed atypical moles cause a dysplastic naevus syndrome to arise which spreads horizontally within the epidermis across the basement membrane shown in Figure 1. If melanocytes are kept in epidermis, it is termed in situ melanoma (Oakley 2012). Vertical growth phase (VGP): With time, malignant melanocytes metastasize and proliferate through the basement membrane into the dermis and subcutaneous tissues. (Oakley 2012). Metastasis: as VGP develops, melanoma becomes thickened and raised; it metastasizes and colonizes to lymph nodes via the lymphatic system or other organ tissues such as the lungs, liver and brain via blood to develop a secondary cancer shown in Figure 2 (Khushiro 2012). This depends on the depth of cell that penetrated the skin (Oakley 2012). 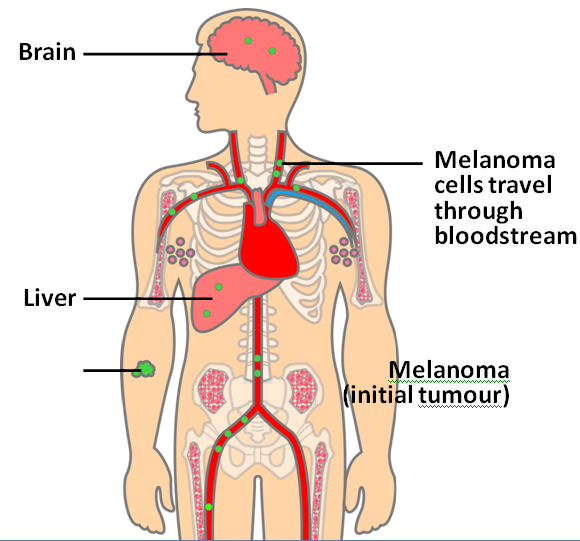
Figure 2: Malignant melanoma (Mee 2012)
Fact box: How is melanoma diagnosed?It begins with examining the mole using ABCDE (Asymmetry, Border, Bleeding, Colour, Diameter, Evolve) checklist to distinguish between a normal mole and melanoma. Normal moles are round, single-coloured whereas most melanomas have multi-coloured, irregular-shaped moles shown in Figure 3. They can be itchy or tender and bleed or crust. Further testing includes biopsies, microscope examination, X-ray, magnetic resonance imaging (MRI) and computerised tomography (CT) and blood tests (British Association of Dermatologists 2011). DNA microarray technology allows identifications of genes involved in tumourgenesis; mRNA that encodes for proteins such as TRP-1, melanoma antigen gp65, monoxcyte chemotactic protein 1 and melanoma differentiating antigen WAF1. With advances in microarray, gene expression regulation that takes place from a normal melanocyte to malignant melanoma can be studied accurately. This indicates how microarray can explain molecular mechanisms of tumour progression and genetic defects involved in MM and holds promises in melanoma care to improve screening methods and develop treatments using gene-directed therapy (Kim 2002). 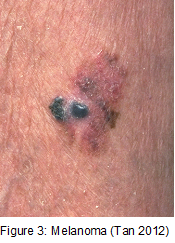
What are melanocytes and how are they normally regulated? Melanocytes are one of the predominant cell types located in the epidermis shown in Figure 4. They produce a pigment called melanin that functions to protect the skin and is responsible for the natural skin colours; this process is called melanogenesis (Cancer Research UK 2011). 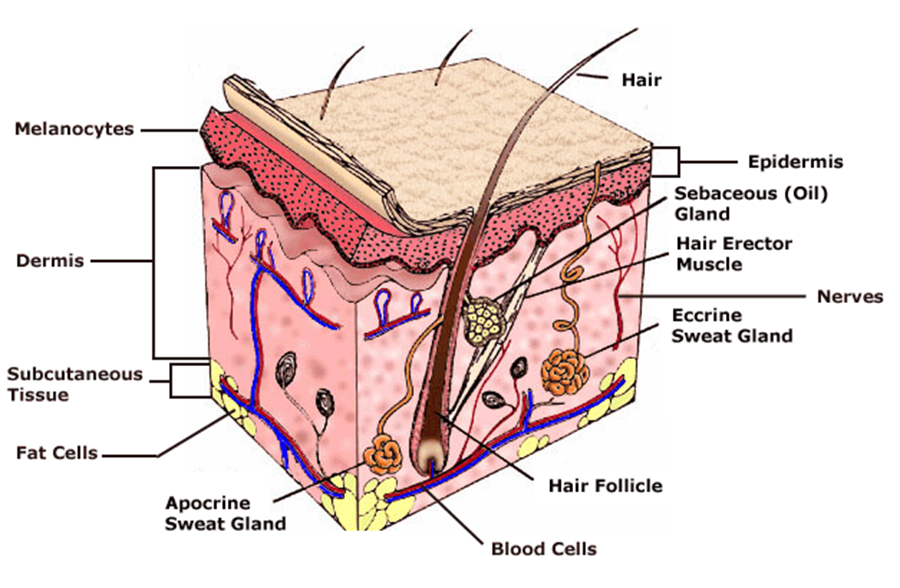
Figure 4: Skin Structure (American Skin Association 2012) The two main cell-signalling pathways that are important in the differentiation, survival and function of melanocytes are RAS mitogen-activated protein kinase (MAPK) pathway and phosphatidylinositol 3 kinase (AKT/PI3K) signalling pathway shown in Figure 5. 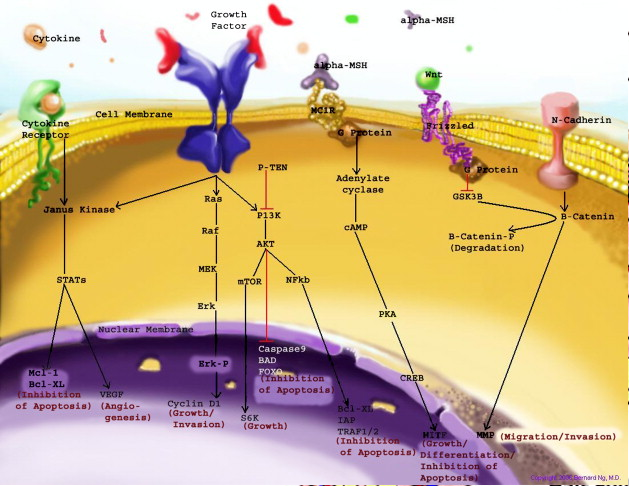
Figure 5: Regulatory processes involved in melanin synthesis (Carlson 2007) Growth factors bind to their specific receptors to activate RAS proteins which sequentially stimulate a phosphorylation cascade of cytosolic protein kinases. It begins with the downstream target called RAF then mitogen-activated protein kinase (MEK1/2) which then acts on extracellular-related kinases (ERK). ERK also interacts with (AKT/PI3K). Phosphorylated ERK kinases are translocated to the nucleus to regulate transcriptional activity of genes that promotes cell cycle progression and proliferation. The PI3K-AKT pathway mediates cell survival signalling via growth factors such as PDGF, NGF, and IGF-1 (Carlson 2007). Phosphatase and tensin homolog (PTEN) is a specific phosphatase that inhibits growth factor signalling by dephosphorylating the plasma membrane lipid phosphatidylinositol 3,4,5-trisphosphate (PIP3) in target proteins to produce phosphatidylinositol 4,5-bis phosphate (PIP2). PIP2 acts as a second messenger and phosphorylates serine/threonine protein kinases AKT. AKT can up-regulate mTOR (mammalian target of rapamycin) for cell growth and S6K, and NFκβ to inhibit apoptosis (Carlson 2007). α-MSH-MC1R (alpha-melanocyte stimulating hormone – melanocortotropin 1 receptor) pathway is also involved in melanocyte regulation. Melanocortins consist of α-MSH and adrenocorticotropic hormone (ACTH) peptides derived from a large precursor protein called pro-opiomelanocortin (POMC). Both MSH and ACTH can bind to MC2R which is coupled to G-protein alpha-s subunit which stimulates adenylyl cyclise (Convanse 2012). Adenylyl cyclase catalyses the cyclization of adenosine triphosphate (ATP) into the secondary messenger cyclic adenosine monophosphate (cAMP) shown in Figure 6. 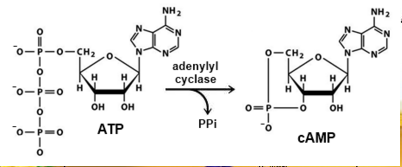
Figure 6: cAMP formation Fact Box: MSH and ACTH MSH is secreted by the intermediate lobe of the pituitary gland and stimulates melanin release and control melanin pigmentation. ACTH is a steroid hormone secreted by the anterior pituitary in response to corticortropin-releasing hormone from the hypothalamus and stimulates the adrenal cortext to secrete cortisol and has slight control over aldosterone; a steroid hormone from adrenal cortex (Bowen 1998). cAMP activates a cAMP-dependent enzyme called protein kinase A (PKA). PKA is a heterotetramer composed of catalytic and regulatory subunits shown in Figure 7. 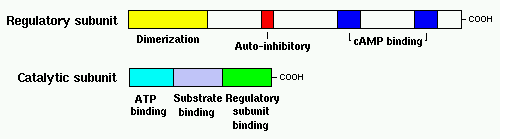
Figure 7: PKA structure PKA, in turn, phosphorylates and activates cAMP response element-binding protein 1 (CREB1). Phosphorylated CREB1 increases microphthalmia-associated transcription factor (MITF) expression in melanocytes, MITF transcription also regulated by Paired box 6 (PAX6) (Convanse 2011). MITF’s three-part structure aids in its function: Basic matif: attaches to specific DNA locations to help control melanocyte development, survival and function. Helix-loop-helix motif and leucine-zipper motif: important for protein interactions as MITF helps control activity of particular genes coding MRPs through interactions with M- and E-boxes located in the promoter regions of Tyrosinase-related protein 1 (TYRP1) and Melan-A (MLANA) (ref, ref). Within normal melanocytes, it also controls melanogenesis and cell cycle arrest whereas in melanoma it holds anti-apoptotic properties. Sp1 transcription factor (SP1) promotes Melanoma cell adhesion molecule (MCAM) expression in melanocytes. Consequently, all of these molecular mechanisms mediate melanocyte differentiation leading to melanin synthesis (Convance 2012) What is the link between obesity and malignant melanoma? The link between obesity and malignant melanoma is not fully understood and a number of studies were performed to investigate whether obesity increases MM incidence. Below are the current understandings of their relationship: Theory One: Leptin resistance Fact Box: What is leptin? It is a protein hormone with a mass of 16 kDa, encoded by the obese gene and is expressed predominantly by adipocytes. Tiny amounts of leptin are secreted in epithelials cells of the stomach and in placenta. It regulates body weight, metabolism and reproductive function. Leptin receptors are expressed in the hypothalamus, T lymphocytes and vascular endothelial cells to regulate body weight (Bowen 2006). Matriese et al. explained that leptin resistance is critical in obesity development and affects leptins’ functions. Most obese individuals have high plasma leptin concentrations that indicate leptin resistance. There are a number of defects that contribute to its resistance: · Defect in leptin transport across blood-brain barrier which reduces leptin’s availability to its receptor. · Defects in leptin receptor signal transduction such as impairment of STAT-3 phosphorylation · Reduced leptin receptor expression · Induction of leptin-signalling inhibitors such as SOCS3 (suppressor of cytokine signalling). Murpurgo et al. suggested that the increased risk of malignant melanoma in obese individuals is primarily due to leptin resistance which highlights the role of leptin in melanoma progression. Specifically, these individuals exhibit reduction of melanogenesis and the ability for melanocytes to repair DNA. Consequently, this increases MSH antagonists shown in Figure 8. All four antagonists lowers MSH activity and MCR 1 and 4, both receptors are members of the five-member G-protein coupled melanocortin receptor family and MCR1 is critical for melanogenesis and melanocyte DNA repair. 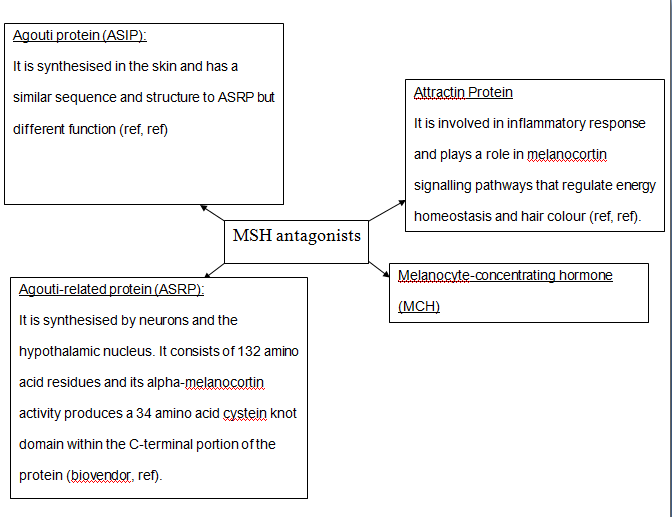
Figure 8: MSH antagonists Theory 2: Cav-1 and FASN involvement Pandey et al. found that diet-induced obesity enhances melanoma growth and was associated with increased Cav-1 and FASN expression. Both are co-ordinately regulated where Cav-1 interacts with FASN in melanoma cells to control proliferation. This demonstrates how diet-induced obesity effects melanoma progression and also indicates how obesity affects critical pathways in melanoma. Cav-1 (caveolin-1) is one of the three integral proteins of the caveolin family. They form hetero- and homooligomers that interact with cholesterol and other lipids to form the sphingolipid-enriched plasma membrane structure. They are involved in cholesterol homeostasis, cell adhesion, apoptosis and interact with a number of signalling molecules for example Gα subunit, tyrosine kinase receptors, PKCs, Src family tyrosine kinases, and eNOS (cell signal). Fatty acid synthase (FASN) catalyses long chains of fatty acids synthesis from acetyl-CoA and malonyl-CoA. It produces lipids in the liver for metabolically-active tissues or storage in adipocytes (cell signal). Theory Three: Melanoma risk is associated with serum leptin levels However, Gogas et al. findings opposes Matriese et al. because they revealed that elevated serum leptin levels in obese individuals was linked to melanoma risk and not with obesity. This result may be due to BMI and adipose tissue may not be one and the same. The major factor that influences serum leptin concentration is adipose tissue mass. Leptin synthesis is affected by several factors: insulin, tumour necrosis factor alpha, glucocorticoids, gonadal hormones, catecholamines and prostaglandins. They also found that: · Leptin expression can be stimulated under hypoxic conditions and often occur in solid tumours. · Leptin connects obesity with melanoma development by stimulating proliferation; it enhances endothelial cell migration and angiogenesis. Leptin stimulates angiogenesis independently and in interaction with two growth factors: vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF-2). In normal angiogenesis, during embryogenesis vasculature development consists of new endothelial cells associated into tubes by the process termed vasculogenesis in addition to sprouting (angiogenesis) of new vessels from existin vessels. Subsequently, normal vasculature becomes quiescent. Similar to normal tissues, tumours need nutrients, oxygen and the ability to excrete metabolic wastes and carbon dioxide. The tumour-associated neovasculature created by angiogenesis address these needs. · Leptin increases endothelial cell growth and suppress apoptosis through a bcl-2 dependent mechanism shown in Figure 9 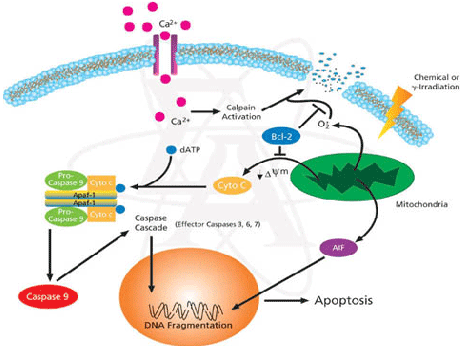
Figure 9: Bcl-2 plays a role in the intrinsic pathway which involves mitochondrial protein release such as cytochrome C interacting with other cytosolic protein factors: apoptoic protease activating factor-1 (Apaf-1) and procaspase-9 to stimulate the association of a caspase-activating complex called a apoptosome. This induces activation of caspase-9 and initiates the apoptotic caspase cascade. By interacting with Bax, Bcl-2 can inhibit pro-apoptotic activity by inhibiting Bcl-2. · Leptin can act as a mitogen (an agent that induces mitosis) or a migration-inducing factor in different cell types such as smooth muscles, normalized and neoplastic colon cells and normal and malignant mammary epithelial cells. Furthermore, Gogas et al. findings needed to be confirmed in future studies to enable the understanding of pathophysiological mechanism and the role of the above factors in predicting melanoma risk. Fact box: What are VEGFs? They are dimeric glycoproteins with a mass of 40kDa and have five forms: VEGFA, VEGFB, VEGFC, VEGFD and PLGF (Mee 2012). Its roles are shown in Figure 10 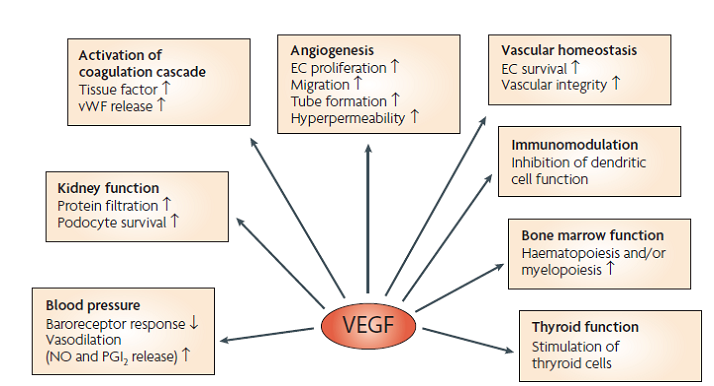
Figure 10: VEGF functions Fact box: What are FGFs? FGFs activates its receptors (FGFRs) on endothelial cells and can induce angiogenic factors from other cell types to indirectly stimulate angiogenesis. Low FGF levels are necessary to maintain vascular integrity. Abnormal FGF signalling enhances tumour angiogenesis and mediates escape of tumour vascularisation from VEGF (Mee 2012). Theory Four: Obesity promotes melanoma progression with or without leptin’s presence Brandon et al. (2009) found limitations in previous studies as their experiments were performed in vitro with high leptin concentrations. They concluded from their mouse studies that the relationship between obesity and melanoma growth was unclear and leptins’ role in promoting melanoma progression is still uncertain. This was due to their following findings: · Obesity promoted melanoma progression whether leptin was present or not. · Energy restriction prevents obesity to reduce tumour size · Leptin deficiency in obesity attenuates progression which correlates with Matriese et al. findings. · Leptin receptors were expressed in melanoma cells but leptin could not be detected as little was produced. However, there is a possibility that leptin interacts with VEGF as their expression was increased in obese mice which suggests obesity increases melanoma growth rate by mechanisms that involve regulating VEGF pathway. However this mechanism does not occur in all cancers. · The leptin found in tumours were derived from host leptin that was removed in the tissues from immune cells that infiltrate the tumour or circulation. Theory Five: Obesity creates a pro-cancerous microenvironment Previous studies suggested that obesity promotes melanoma development by creating a pro-cancerous microenvironment consisting of high levels of growth and inflammatory factors: cancer-associated fibroblasts (FACs), macrophages (TAMs), and adipocytes (Kushiro 2012). Adipocytes secrete inflammatory cytokines that recruit macrophages to initiate angiogenesis by secreting matrix metalloproteinases (MMPs) collectively known as matrixins are calcium-dependent proteases which are synthesizes as a inactive proenzymes and are activated by cleavage of a pro-petide. It hydrolyses extracellular matrix (ECM) and basement membranes of neighbouring blood vessels (Kushiro 2012). MMPs 2 and 9 specifically degrades collagen type IV component of basement membrane. This enables metastasising tumour cells to move through ECM (Mee 2012). A discovery! Recently, miR-126, a member of small non-coding RNA (microRNAs) was revealed to inhibit endothelial recruitment by suppressing a set of cancer genes that activate endothelial MMP is controlled by an increased expression on a transcriptional level. Thus, microRNAs expressed by cancer cells could shape the tumour and metastatic microenvironment (Mee 2012) Theory 6: Obesity increases metastasis by promoting a mesenchymal cell phenotype The Epithelial-to-Mesenchymal Transition (EMT) normally functions in the formation of the body plan and differentiation of multiple tissues and organs and contribute to tissue repair (Mee 2012). It consists of an epithelial-type cell that obtains a mesenchymal phenotype which gives it migratory and invasive ability needed for cancer progression and higher metastatic ability (Khushiro 2012). The mesenchymal phenotype is associated to cells’ ability to migrate to distant organs allowing differentiation into multiple cell types during metastasis initiation and development. EMT in cancer progression is characterized by activation of transcriptional factors such as TGF-β1, Snai1, Twist and Slug abnormally. They aid in inhibiting cell-to-cell adhesion that maintains cell attachment to each other by reducing E-cadherin, an anchorage protein that increases expression of mesenchymal markers such as Vimentin shown in Figure 11and increase invasiveness of tumour cells (Kushiro 2012). Khushiro 2012 found obesity may increase the metastatic ability of melanoma by promoting a mesenchymal cell phenotype. This finding was revealed after finding: · high levels of resistin, insulin, tPAI1, IL-6, TNF-α, and MCP-1 in obese serum which increases invasive ability of melanomas. · They also found that ob/ob serum increased the expression of Snai1 and Twist. Both Snai1 and Twist are strongly associated with EMT and metastasis of melanomas. It increased MMP9 activity and decreased the expression of E-cadherin and the metastasis suppressor gene Kiss1. 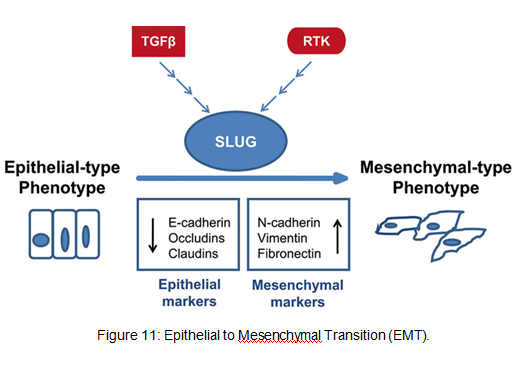
Treatment for obesity and obesity-linked cancers? Over 120 different types of anti-obesity drugs have been tested in clinical trials but only Orlistat has been proved to be safe and effective. It blocks the action of a protein used to digest fat. The undigested fat is passed out with stools to avoid gaining weight but it will not necessarily cause one to lose weight. However, currently approved anti-obesity drugs show only limited efficacy, generally facilitating no more than a 5–10% reduction of body weight and are often associated with side-effects. A new therapeutic target called Gastric inhibitory peptide (GIP) may hold promising novel therapy for long-lasting obesity treatment and also show that vaccination against GIP is safe and effective after recent studies have shown that GIP receptor-deficient mice are finally protected from diet-induced obesity. Normally, GIP promotes triglyceride clearance from the circulation which is partially mediated by its ability to stimulate lipoprotein lipase activity. However prospective pre-clinical safety studies is necessary before the therapy can be introduced in humans (ref, ref) In relation to anti-cancer therapy, researchers from the University of Manchester (2008) recommended national cancer plans that entail the following to reduce obesity, decrease cancer incidence and increase survival: · Restriction for high-calorie and low-nutrient food advertisements · Limited access to unhealthy foods · Promote physical activity in schools and workplaces. · Avoiding alcohol as it contains high calories which increase weight. New Discovery! A yellow compound called Curcumin mediates anticancer and obesity activities but are not yet been publicized. It modulates multiple molecular targets and reverses insulin resistance as well as other symptoms that are associated with obesity-related cancers. Curcumin mediates multiple molecular pathways, and is considered to be of therapeutic value in the treatment and prevention of obesity-related cancers. Conclusion Overall, to summarise what studies have shown about the link between obesity and malignant melanoma:
· Obesity increases metastatic ability of melanoma by enhancing Cav-1 and FASN expression. · It creates a pro-cancerous microenvironment of growth and inflammatory factors to initiate angiogenesis. · Leptin resistance and its high levels in serum in obese individuals. · However, Brandon et al. argues the previous finding and found that obesity promoted melanoma progression whether leptin was present or not. Thus, current molecular mechanisms of how obesity associates with melanoma development and progression is unclear and ongoing research is necessary to understand melanoma pathogenesis in order to establish effective therapeutics with less side effects. Obesity may have a causative relationship with malignant melanoma but it is not the main risk factor. Gogas et al. reported that MM incidence in Caucasian individuals is increasing globally making it the most increasing cancer in white populations except for lung cancer among women. Dark-skinned people have a lower risk because their melanocytes have more melanin thus more natural protection, despite having the same amount of melanocytes as Caucasian people. Other factors include the ‘sensitive’ skin phototype (fair skin, easily to burn and unable to tan), dysplastic nevi presence, family history, previous melanoma and immunosuppression (Cancer Research UK 2011). 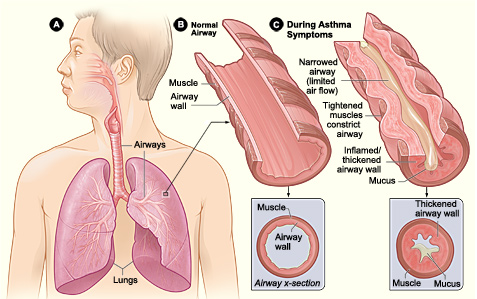 Asthma is an inflammatory disease of the airways; affecting 5.9 million people in the UK (Asthma UK 2011). There are two asthmatic forms: Intrinsic and Extrinsic. Extrinsic asthma affects children and over 80% of its deaths occur in developing countries. However, it can occur anywhere regardless of development levels (WHO, 2011). Intrinsic asthma affects elderly. Amongst the common symptoms of asthma are wheezing, coughing and dyspnoea (breathlessness). Dry coughing associated with wheezing due to bronchoconstriction and causes sleep disturbances particularly in the early hours (Munro, 2000:118). These clinical features are the outcome of a complex cascade of events in the inflammatory mechanism proceeding, including structural and biological changes after her airways were exposed to a stimulus. For instance, dust. Normally, inhaled antigens are eliminated by mucociliary escalator mechanisms in the tracheobronchial tree. The escalator consists of mucous-producing goblet cells and ciliated epithelium. It removes particles that sediment the airways by cilia continuously beat and transfers adhesive mucus containing antigens out and up the larynx to the epiglottis to be swallowed (University of Colorado, 2011). Asthma is diagnosed via taking a medical history and breathing tests to ensure how well lungs are working. A spirometry is a test where you take a deep breath and blow into a sensor. This test measures the amount of air the lungs is needed to hold and the speed of the air for inhaling (breathing in) and exhaling (breathing out. It ensure examines how severe the asthma is and whether the treatment is working. An allergy test may also take place to see what triggers your asthma. This could be animal fur, dust, pollen, nut or something that will trigger your asthma. Thus, identifying what triggers your asthma will aid in helping to manage your asthma. Is there are treatment for asthma? Symptoms experienced can be controlled using effective treatment and management.Amongst are the following: leukotriene modifiers: These are oral medications that help relief quickly symptoms and helps relax and open the airways. Controller medications: These are taken daily. Examples include inhaled corticosteroids such as beclomethasone, fluticason, mometasone. Combination inhalers: This consists of inhaled corticosteroids and a long-acting beta-agonist. LABAs controls the symptoms and opens the airways. However, it has side-effects and thus it is never prescribed solely for asthma and thus is used alongside corticosteroids. Examples of combination inhalers are (salmaterol and fluticasone). Oral and intravenous corticosteroids – This is needed for severe symptoms. Examples include prednisone. 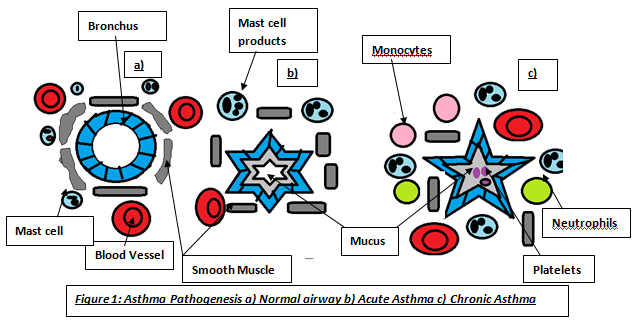 WANT TO DIP DEEP INTO ASTHMA?The pathophysiology of asthma is divided into early and late phase reactions. The early phase causes a forced expiratory volume in 1 second, whereas, in the late phase causes a decrease in bronchial airflow for 4-8 hours (Fireman, P. n.d.). The inflammatory cells responsible for most clinical symptoms are mast cells, basophils, eosinophils and lymphocytes. The inhaled allergens penetrate the respiratory epithelium and are captured by dendritic cells. The antigens are presented to the B and T lymphocytes. The B-lymphocytes produce Immunoglobulin E (IgE) which is dependent on CD4 T-cells (Price 2004:14). This is immediate hypersensitivity. IgE is a monomer consisting of two heavy chain containing four C-domains, and two light chains (Wood 2001:47). The cells differentiate into Th2 cells secreting interleukin-4 (IL-4) responsible for allergen-specific IgE production. The Fc part of these allergens bind to the F¢εR receptors on membrane surfaces of mast cells (Wimberly 2001:148). Mast cells are responsible for the initial inflammatory response due to their residence in tissues; however, there are two types with different properties: mucosal found in the lungs, whereas, connective found in most tissues (Wood 2001:233). This binding activates and degranulates mast cells releasing mediators into the blood. There are two types of anaphylactic mediators: Preformed stored in granules of mast cells and basophils; histamine and enzymes. Newly-generated mediators produced after mast cell activation; arachidnoic acid metabolites. Phospholipase releases arachidonic acid from phospholipids and are exposed to enzymes making metabolites: LTC4 and PGD2. These mediators cause Ruby’s allergic manifestations and can cause acute asthma attacks.
The late phase is due to influx of other inflammatory cells that further stimulate mucus secretion, epithelial cell damage and activation of platelets leading to microthrombi in the airway (Wood 235), characterized by airway inflammation leading to epithelial denudation, cellular infiltration, submucosal oedema and increased collagen deposition under the epithelial basement membrane. An increase in collagen also produces sub-basement fibrotic effects which remodels airway with irreversible obstructions (Clark 1998:9). Microscopically, asthma is characterized by the presence of increased numbers of eosinophils, neutrophils, lymphocytes, and plasma cells in the bronchial tissues, bronchial secretions, and mucus. Four events during inflammatory response|: vasodilation cause increase blood flow, increase supply of cells...activation of endothelial cells making them more sticky to blood cells, increase vascular permeability-cells and proteins pass through vessel wall. Mast cells also release enzymes that degrade blood vessel endothelial membrane. Tryptase, a serine protease has high abundance and are used in studies regarding asthmatic response evaluation. Its large structure allows slow movement around the inflammation site and enhances histaminic effects by: basement membrane thickening. It also degrades both bronchoactive peptides and vasoactive intestinal peptide that inhibits smooth muscle relaxation and causes over-proliferation of epithelium respectively. It can act as an anticoagulant transporting inflammatory cells to the injury site. These roles cause significant structural changes of the airway and bronchoconstriction. Wheezing are sounds produced by air passage through narrowed bronchi (Munro 123) wheezing is louder during expiration and often confided . it is more conspicuous and audible only during deep breathing.. patients with stridor can also get wheeze. Care needs to be distinguished between two sounds as stridor is caused by partial obstruction of a major airway (e.g laryngeal oedema, tumour and needs urgent treatment (Munro 123) Another enzyme is chymase which is not abundant as tryptase but have similar roles. For instance, neuroactive peptides degradation and vascular permeability increasing inflammation as serum proteins and fluid travel across endothelium. Matrix proteins degradation causes epithelial damage (Wimberly 2001:149). Basophils can bind to IgE. Basophils and IL-3 originates in the bone marrow stimulating cell differentiation and binds to IgG. Its granules contain histamine and IL-4. Histamine is produced via histidine decarboxylase. Its effects on lung tissue depend on receptor stimulated. The H1 receptor on smooth muscle causes muscle contraction leading to bronchoconstriction and airway oedema (Wimberly 2001:149). This caused Ruby’s prolonged breathless at hospital. Increases work of breathing, increase ventilator drive and imapaired respiratory muscle function (Jamison 120) This condition is called bronchospasm. Eosinophils invade the bronchial wall resulting in airway becoming oedematous (Clark 1998:9). The H2 receptors causes yellow, viscid and regulates histamine release via negative feedback. Mucus hypersecretion can lead to plugging due to mucus and desquamated epithelial cells and the sputum can be coughed up in acute attacks (Clark 1998:9). Other manifestations include watery eyes and nasal discharge. Another histaminic role is antibody and prostaglandin hypersecretion. Prostaglandin (PGD2) is a bronchoconstrictor and can also be synthesized by cyclooxygenase pathway (COX) to make prostaglandins and thrombaxane b2 Another bronchoconstrictor is LTC4 a leukotriene produced by 5-hydroperoxyeicosatetraenoic acid in lipoxygenase. LTC4 becomes a contractile agonist at the CisLTs receptor, located on the bronchial smooth muscle resulting in bronchoconstriction. (Wimberly 2001:149). Tumour necrosis factor-α, IL-4, IL-5, IL-6 are cytokines are important in asthmatic attack propagation. TNF-α is secreted in mast cells and increases adhesion molecule levels (selectins – bind to sugars on mucins, integrins-bind to ICAMScausing leukocytes activation and adhesion of: eosinophils, basophils and neutrophils to endothelium and migrate to the injury site promoting inflammation. Besides IgE synthesis, IL-6 is involved in reducing cytokine effects. IL-5 involves eosinophil production abundant in alveolar lavage fluid. For eosinophil and macrophage maturation, T –cells release IL-3, IL-5 and granulocyte-macrophage colony-stimulating factor (GM-CSF). These cells are critical for prolonged asthmatic response due to acculumation of these leads to epithelial denudation and airway oedema they are inactivated form and lose granules after allergen clark p102. Denudation is caused by major basic protein (MBP) located in eosinophil granules, whereas, oedema results from epithelial damage from MBP. Epithelium produce epithelin which is a bronchoconstrictor and vasoconstrictor, nitric oxide is a vasodilator and bronchodilator. NO production may be stimulated by cytokines and NO produced has cytoxic effects on epithelium (clark 102). Bronchoconstriction may be caused by MBP enhancing effect of cholinergic contraction of airways. Cholinergic effects on lungs via acetylcholine cause bronchoconstriction, mucus hypersecretion and bronchial vasodilation. If airways are not hyper-responsive, cholinergic effects maintains airway tone through vagus nerve and increases vagal output which normally occurs during stress or at night. This emphasises why she was breathless when she was distressed. Chonlinergic fibres constrict smooth muscle, stimulation of beta adrenoreceptors dilates muscle, little adrenergic nerve supply bronchials smooth muscle so adrenaline provides main stimulus to thesr receptors. Third system is nonadrenergic, noncholingeric system that has transmitters that produce constriction or dilation, inflammatory mediators may affect release of neutrotransmittors (clark 103)Besides these cells, studies have shown that chemotactic factors such as eotaxin have shown to be eosinphil chemoattractants and eosinophils promote pathophysiological dysfunction including peribronchial thickening and fibrosis (Nature, 2011) Studies show that lymphocytes are important in orchestration of inflammatory response rather than sources of mediators (Clark 102). Macrophage originates from blood monocytes and must be present for T-cell activation and secrete cytokines and chemotactic factors all involved in asthmatic airway response clark 102. This occurs through a signal passed between the B7 molecule located on macrophage and the CD28 molecule on the T cell. For instance, IL-1, IL-6 and TNF-β. IL-1 enhances T and B-cell activation and increase capillary permeability allowing inflammatory components to migrate neutrophil/polymorphonulear leukocyte to injury site increasing oxidant levels. Oxidants potentiate the airway causing epithelial damage, bronchoconstriction, and decreased ciliary action leading to airflow obstruction. IL-1 causes an increased production of other cytokines. TNF-β stimulates cytokine production and increase endothelial permeability to facilitate blood cells movement to injured site. Do you carry a bottle of water with you? If not; I think after when you finish reading this article you probably will carry one! So what are the benefits of drinking water? A) Helps control calories! Worried about how much calories you eat or drink? Water does not have any magical effect by making one lose weight. However it is a good subsititute than having fizzy or soft drinks. Eating food that has a high water content such as fruits, vegetables, soups will require more chewing. Therefore, the body absorbs it more slowly and so it will help you feel full. B) It helps make your skin healthy and good! Really? How?! Well the skin consists of plenty of water and acts as a protective barrier to prevent excess water loss. Dehydration makes your skin look dry, which can be improved with hydration. However, once adequately hydrated, the kidneys take over and excrete excess fluids." Using a moisturizer also helps to keep the moisture in the skin. C) It helps maintain the bowel function! It prevents constipation. Constipation is when there is not enough fluid in your digestive system; so your large intestine pulls water from the stools/number 2 to maintain hydration resulting in constipation. D) It helps maintain a balance of body fluids! Our bodies are composed of 60% water. The roles of body fluids is to maintain body temperature, make saliva, digestion, circulation and transport nutrients. When you're low on fluids, the brain triggers the body's thirst mechanism. Alcohol can damage the brain and liver. It can also lead to excess excretion of fluids which leads to dehydration. E) It helps give energy to muscles! When there is an imbalance of fluides and electrolytes. This can lead to muscle fatigue. F) It helps your kidneys! Bodily fluids helps transport waste products such as toxins like urea to be excreted in the urine using the kidneys. Thus in order to keep the kidneys in function, there needs to be an adequate intake of fluids. When you are drinking a good amount. The colour of your urine is light and has no smell. However, if you are not drinking a good amount, your urine will become concentrated and have a darker shade of colour as well as a smell. This is due to the kidneys are trapping extra fluid to keep the bodily functions going. Drinking little water can increase the risk of kidney stones.!   |
This project began as a facebook page sharing information about different illnesses, diagnosis and treatments. We are now doing short articles :)
Health stuffArchives
April 2020
Categories |
 RSS Feed
RSS Feed
